



Zarządzanie Ryzykiem w Cyklu Życia Walidacji cz. 2
Po aktualizacji Aneksu 15 ‘Kwalifikacja i Walidacja’ przez europejską agencję EMA, integralną częścią programu walidacji staje się zarządzanie ryzykiem jakości w trakcie całego cyklu życia produktu. Wdrożenie formalnego zarządzania ryzykiem wymaga opracowania metodologii, umożliwiającej ocenę krytyczności atrybutów jakościowych produktu i parametrów procesu oraz okresową ocenę ryzyka związanego z jakością produktu leczniczego.

Aneks 15 Dobrej Praktyki Wytwarzania, zawierający zasady kwalifikacji i walidacji, został zaktualizowany przez europejską agencję EMA w 2015 r., wprowadzając wytyczne Międzynarodowej Konferencji ds. Harmonizacji ICH Q8, Q9 i Q10. Nowe podejście do walidacji zakłada stosowanie formalnego zarządzania ryzkiem jakości, w trakcie cyklu życia produktu, jako narzędzia do oceny krytyczności i analizy ryzyka jakościowych atrybutów produktu oraz parametrów procesu. Istotą wprowadzonych zmian jest systematyczne budowanie wiedzy o procesie i produkcie w trakcie cyklu życia produktu i ciągłe doskonalenie procesu poprzez identyfikację, redukcję i kontrolę źródeł zmienności. Ogólne zasady zarządzania ryzykiem przedstawione zostały w części I artykułu, część II zawiera opis wybranych metod zarządzania ryzykiem z przykładami, które mogą być wykorzystane w programie walidacji do oceny krytyczności i szacowania ryzyka.
Parametry i atrybuty
Zgodnie z wymaganiami zawartymi w Aneksie 15 dokumentacja walidacyjna powinna zawierać listę krytycznych parametrów procesu (CPP Critical Process Parameters) oraz krytycznych atrybutów produktu (CQA Critical Quality Attributes), wraz z podaniem uzasadnienia odnoszącego się do prac badawczo-rozwojowych i udokumentowanej wiedzy o procesie. Aneks 15 i dokumenty ICH nie precyzują jednak pojęcia krytyczności, ani nie podają metodologii, która mogłaby być wykorzystana do zdefiniowania granicy pomiędzy tym, co krytyczne a niekrytyczne w procesie. W zakresie odpowiedzialności wytwórcy pozostaje więc zdefiniowanie znaczenia pojęcia „krytyczny”, jak również wybór metodologii definiowania krytyczności i oceny ryzyka. Każda metodologia powinna być zgodna z definicjami krytycznego atrybutu i parametru procesu, zawartymi w aneksie 15. Jednak podział parametrów na krytyczne i niekrytyczne prowadzi do dwóch skrajnych możliwych rozwiązań. W jednym wszystkie parametry można uznać za krytyczne, ponieważ muszą spełniać założone limity, a tym samym powinny być kontrolowane, a w drugim żaden parametr nie jest krytyczny, gdyż jest kontrolowany i musi spełnić założone kryteria. Oba podejścia wydają się niepraktyczne. Konieczne jest więc wypracowanie rozwiązania, które odpowie na pytania:
- Czy krytyczność powinna być oparta na podziale krytyczne / niekrytyczne czy raczej stanowić kontinuum, zaczynając od wysokiej poprzez umiarkowaną do niskiej?
- Jak zdefiniować granice między wysoką, umiarkowaną a niską krytycznością?
- Czy krytyczność atrybutów jakościowych ulega zmianie w cyklu życia produktu ?
- Czy krytyczność parametrów procesu ulega zmianie w cyklu życia produktu?
- Jak powiązać krytyczność jakościowego atrybutu produktu i parametru procesu z oceną ryzyka?
- Jak powiązać ocenę krytyczności i ryzyka z pozostałymi programami systemu zapewnienia jakości?
Procedury przedstawione na schemacie, obejmujące ocenę wiedzy o produkcie, ocenę poznania procesu oraz ocenę strategii kontroli procesu, odpowiadając na postawione pytania, mogą być wykorzystane w analizie krytyczności i ryzyka (rys.1).
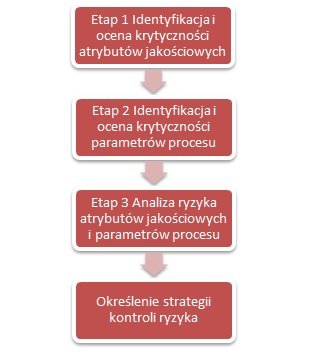
RYS. 1 Schemat oceny krytyczności i ryzyka
Ocena krytyczności atrybutów jakościowych
Celem metody jest identyfikacja oraz ocena krytyczności atrybutów jakościowych produktu. Krytyczne atrybuty produktu, to: fizyczne, chemiczne, biologiczne, mikrobiologiczne własności i charakterystyki, które powinny być zgodne z zatwierdzonym limitem, zakresem lub rozkładem w celu zapewnienia wymaganej jakości produktu. Krytyczność atrybutu jakościowego, będąc pochodną szkodliwości, nie ulega zmianie w trakcie cyklu życia produktu.
Metoda może mieć zastosowanie w cyklu życia walidacji od etapu 1 projektowania procesu, poprzez etap 2 walidacji procesu, a następnie podczas etapu 3 bieżącej weryfikacji procesu.
Dla nowo rozwijanych produktów należy ustalić atrybuty jakościowe na podstawie zdefiniowanego docelowego profilu jakości produktu (QTPP Quality Target Product Profile).
Dla produktów istniejących można zdefiniować atrybuty jakościowe, wykorzystując specyfikacje produktu gotowego oraz produktów pośrednich.
Ocena krytyczności
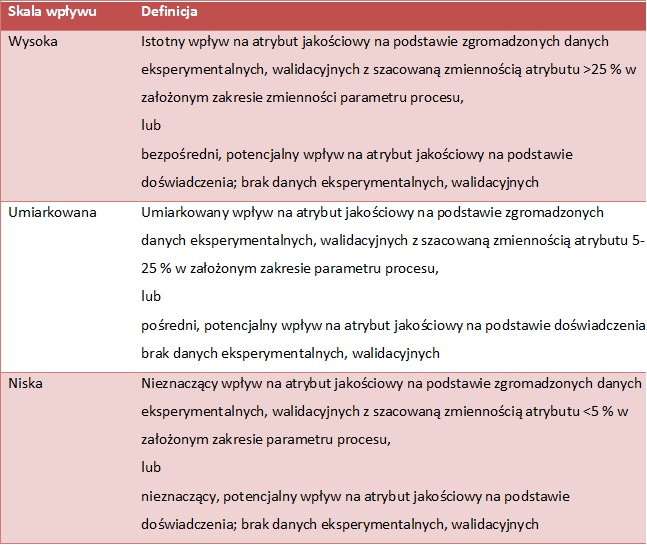
Po zidentyfikowaniu atrybutów jakościowych produktu należy ustalić ich krytyczność biorąc pod uwagę wpływ zmienności parametru na bezpieczeństwo stosowania oraz skuteczność produktu leczniczego zgodnie ze skalą szkodliwości przedstawioną w tab. 1. Podczas szacowania wpływu zmienności atrybutu jakościowego na życie i zdrowie pacjenta powinien być uwzględniony stopień niepewności oceny, wynikający z niedostatecznej wiedzy, luk w naukach farmaceutycznych i zrozumieniu procesu. Ocena krytyczności może być przedstawiona w tabeli z podaniem nazwy atrybutu jakościowego produktu, przypisanego poziomu krytyczności oraz uzasadnienia z odniesieniem do źródła wiedzy: dane eksperymentalne, dane literaturowe lub doświadczenie. (tab. 2)

TAB. 1 Skala szkodliwości atrybutu jakościowego
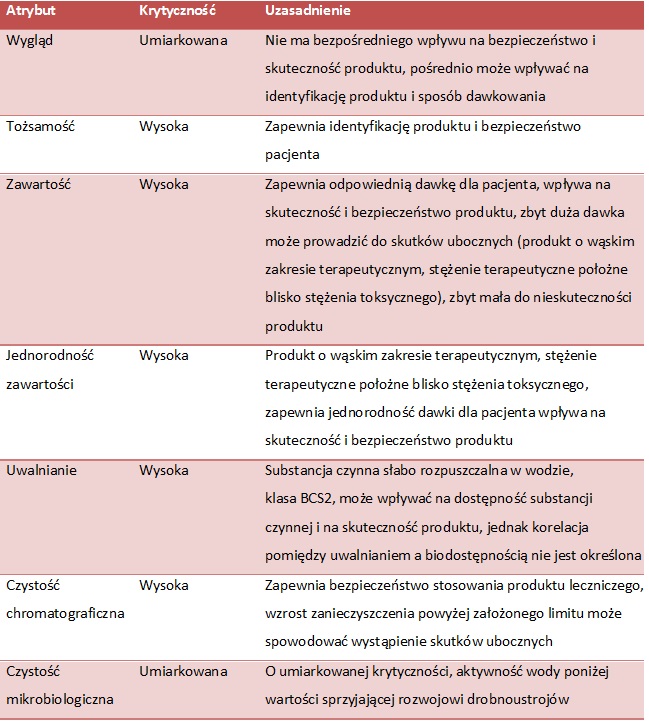
TAB. 2 Ocena krytyczności atrybutów doustnego produktu leczniczego
Ocena krytyczności parametrów procesu
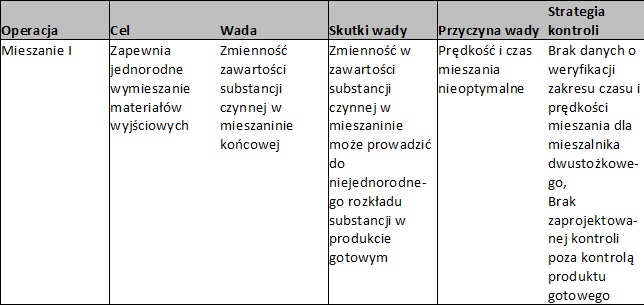
Celem metody jest identyfikacja oraz ocena krytyczności parametrów procesu (źródeł zmienności) dla każdej operacji jednostkowej procesu. Krytyczny parametr procesu to parametr, którego zmienność może mieć wpływ na krytyczny atrybut jakościowy i który powinien być monitorowany lub kontrolowany w celu zapewnienia, że proces wytwarza produkt o wymaganej jakości. Krytyczność operacji jednostkowej/parametru procesu, będąc kombinacją szkodliwości atrybutu jakościowego i potencjalnego wpływu parametru procesu, może ulegać zmianie w trakcie cyklu życia produktu w miarę gromadzenia wiedzy o produkcie i procesie.
Metoda może mieć zastosowanie w cyklu życia walidacji od etapu 1 projektowania procesu, poprzez etap 2 walidacji procesu a następnie podczas etapu 3 bieżącej weryfikacji procesu.
Identyfikacja operacji jednostkowych
Pierwszym krokiem jest zdefiniowanie operacji jednostkowych procesu, podczas których materiały/produkt pośredni są chemicznie lub fizycznie przetwarzane/przechowywane. Identyfikację operacji można przedstawić za pomocą diagramu przepływu procesu, podając materiały wyjściowe, typ stosowanego urządzenia oraz punkty kontroli międzyoperacyjnej/produktu gotowego na poszczególnych etapach procesu (rys.2).
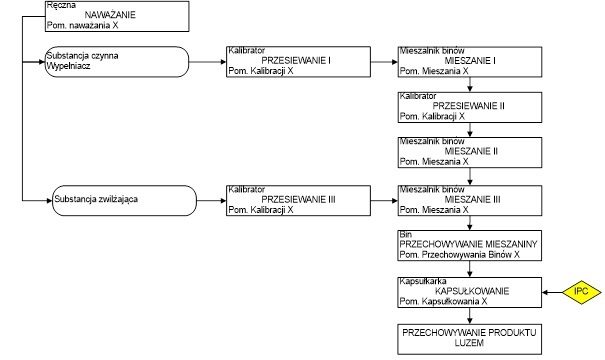
RYS. 2 Diagram przepływu procesu
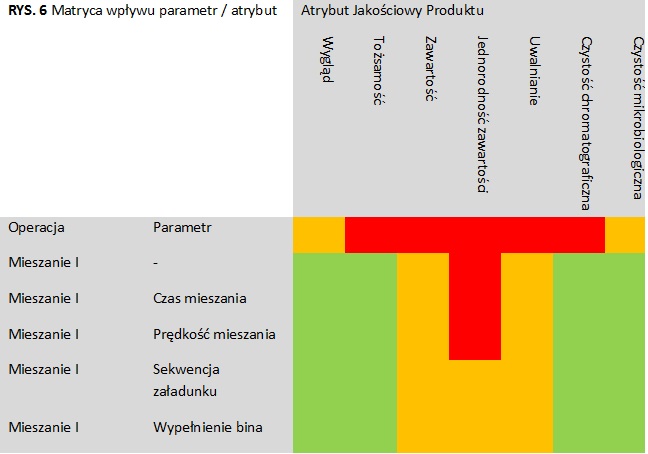
Kolejnym etapem jest zdefiniowanie parametrów odpowiedzi procesu dla każdej operacji jednostkowej, włączając atrybuty produktów pośrednich i produktu gotowego. Następnie należy ustalić parametry procesu dla każdej operacji jednostkowej, uwzględniając parametry operacyjne urządzeń, parametry materiałów wyjściowych, czas i warunki przechowywania produktów pośrednich, parametry środowiska oraz parametry odpowiedzi procesu wynikające z poprzedniej operacji. Identyfikację parametrów można przedstawić za pomocą diagramu przyczynowo skutkowego (rys. 3).
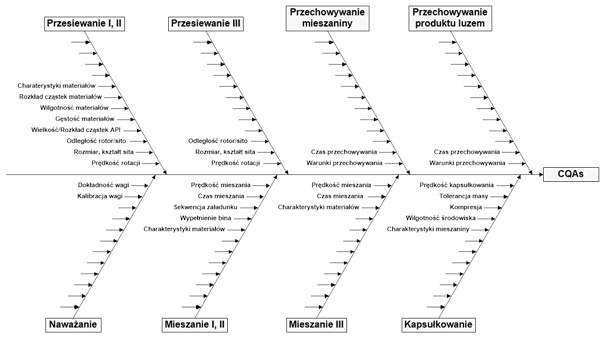
RYS. 3 Identyfikacja parametrów. Diagram przyczynowo skutkowy
Krytyczność i poziom wpływu
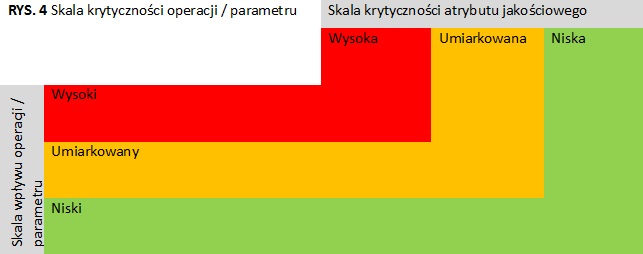
Po zidentyfikowaniu operacji jednostkowych/parametrów procesu, następnym krokiem jest ocena ich potencjalnego wpływu na atrybuty jakościowe produktu zgodnie ze skalą wpływu podaną w tabeli (tab.3). Ocenę wpływu można przedstawić z formie matrycy, wiążącej operację jednostkową/parametry procesu z atrybutami jakościowymi produktu, zaznaczając krytyczność atrybutu oraz poziom wpływu (rys 5, 6). Następnie należy dokonać oceny krytyczności danej operacji/parametru zgodnie ze skalą krytyczności (rys. 4). Biorąc pod uwagę kombinacje szkodliwości atrybutu jakościowego i wpływu parametru procesu na atrybuty jakościowe, wszystkie operacje/parametry można podzielić na trzy kategorie:
- O wysokiej krytyczności (czerwony obszar)
- O umiarkowanej krytyczności (żółty obszar)
- O niskiej krytyczności (zielony obszar)
Poziom wpływu / krytyczności każdego parametru procesu należy odpowiednio uzasadnić, podając źródło wiedzy: dane eksperymentalne, doświadczenie (tab. 4).
TAB. 3 Skala wpływu parametru procesu na atrybuty jakościowe

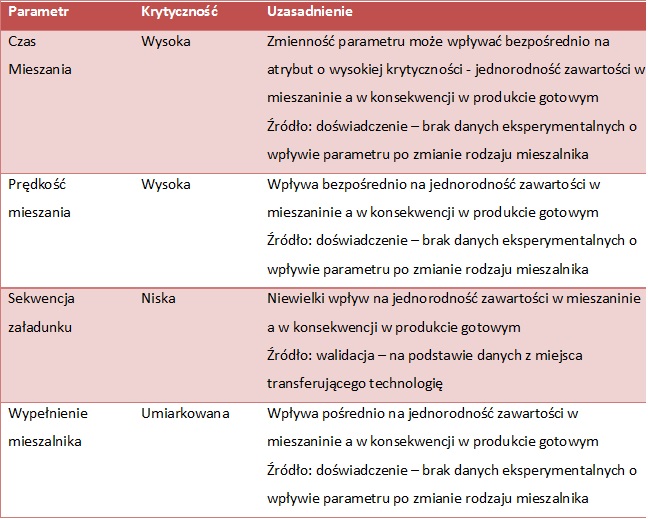
TAB. 4 Ocena krytyczności parametrów procesu
Analiza ryzyka procesu
Celem analizy ryzyka procesu jest identyfikacja ryzyka stwarzanego dla jakości produktu i procesu, a wynikającego z niedostatecznej wiedzy, lukach w naukach farmaceutycznych i zrozumieniu procesu, źródłach szkody (np. przyczyny wad w procesie, źródła zmienności ) oraz prawdopodobieństwa wykrycia problemów. Metoda może mieć zastosowanie w cyklu życia walidacji od etapu 1 projektowania procesu, poprzez etap 2 walidacji procesu a następnie podczas etapu 3 bieżącej weryfikacji procesu.
Analiza ryzyka procesu odbywa się w następujących krokach:
- Krok 1 Identyfikacja Operacji
Na podstawie schematu przepływu procesu, zidentyfikować operacje jednostkowe procesu.
- Krok 2 Identyfikacja Celu Operacji
Dla każdej operacji jednostkowej procesu zdefiniować jej cel.
- Krok 3 Identyfikacja Wad / Zagrożeń / Błędów
Zidentyfikować wady, które mogą spowodować, że cel operacji nie zostanie zrealizowany.
- Krok 4 Identyfikacja Skutków Wad
Dla każdej wady zidentyfikować szkodliwość w odniesieniu do atrybutów jakościowych produktu.
- Krok 5 Identyfikacja Przyczyn Wad
Na podstawie diagramu przyczyno-skutkowego określić potencjalne przyczyny / źródła zmienności wady.
- Krok 6 Identyfikacja Metod Kontroli Wad
Dla każdej wady zidentyfikować metody kontroli procesu, zaprojektowane w celu detekcji, redukcji lub wyeliminowania przyczyn i/lub wad.
- Krok 7 Określenie Ryzyka
Dla każdej wady dokonać oceny:
- szkodliwości wady na podstawie skali szacowania krytyczności atrybutu jakościowego (tab. 1);
- prawdopodobieństwa wystąpienia wady na podstawie skali szacowania wpływu (tab. 3);
- wykrywalności wady na podstawie skali szacowania detekcji przyczyny i/lub wady (tab. 5).

TAB. 5 Skala wykrywalności
- Krok 8 Klasyfikacja Ryzyka
Dokonać klasyfikacji ryzyka, korzystając z oceny krytyczności parametru zgodnie ze skalą krytyczności (rys. 3), wiążąc krytyczność parametru z ryzykiem:
- o wysokim ryzyku (krytyczności) (czerwony obszar),
- o umiarkowanym ryzyku (krytyczności) (żółty obszar),
- o niskim ryzyku (krytyczności) (zielony obszar).
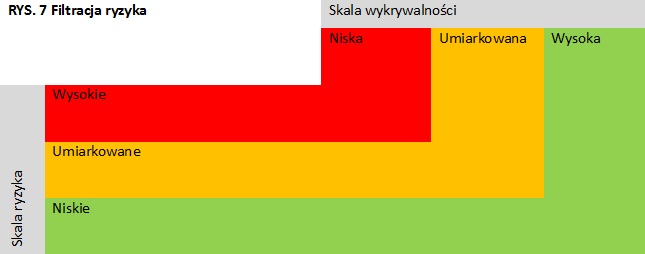
- Krok 9 Filtracja Ryzyka
W oparciu o klasyfikację ryzyka i wykrywalność, przeprowadzić filtrację ryzyka, nadając priorytet zdarzeniom zgodnie ze skalą filtracji ryzyka (rys. 6):
- o największym priorytecie (czerwony obszar),
- o średnim priorytecie (żółty obszar),
- o niskim priorytecie (zielony obszar).
- Krok 10 Akceptacja Ryzyka
Po określeniu ryzyka podjąć decyzję o jego akceptacji. Jeżeli ryzyko jest akceptowalne (obszar żółty lub zielony), proces może pozostać bez zmian lub można podjąć działania w celu dalszej redukcji ryzyka i poprawy procesu. Zagrożenia o nieakceptowanym poziomie ryzyka (obszar czerwony) i o najwyższym priorytecie (obszar czerwony) muszą podlegać procedurze redukcji ryzyka.
- Krok 11 Redukcja Ryzyka
Jeżeli ryzyko jest nieakceptowane, zdefiniować odpowiednie działania naprawcze / zapobiegawcze w celu modyfikacji procesu tak, aby zredukować prawdopodobieństwo wystąpienia przyczyny/wady i/lub zwiększyć poziom kontroli. Preferowana jest redukcja prawdopodobieństwa wystąpienia nad zwiększaniem poziomu kontroli oraz stosowanie rozwiązań technicznych niż proceduralnych.
- Krok 12 Okresowa Ocena Ryzyka
Po zdefiniowaniu działań naprawczych / korygujących lub wprowadzeniu zmian w procesie przeprowadzić ponowną ocenę ryzyka w celu określenia czy ryzyko zostało zredukowane do akceptowalnego poziomu. (tab. 6, 7, 8)
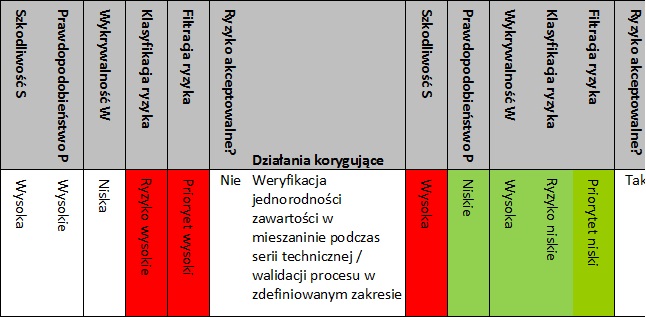
TAB. 6 Analiza ryzyka
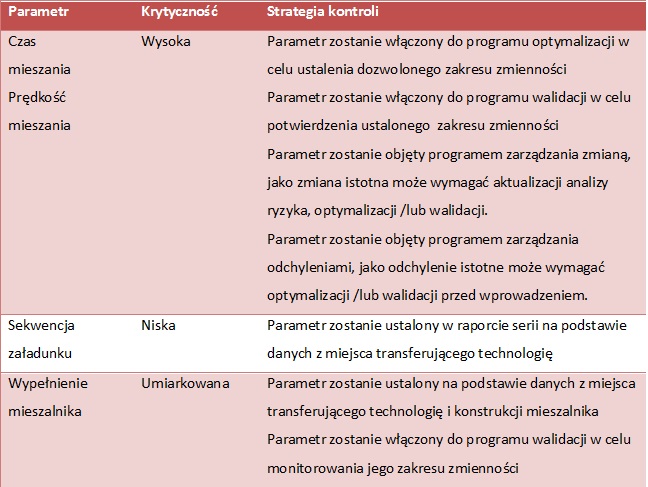
TAB. 7 Ocena strategii kontroli procesu
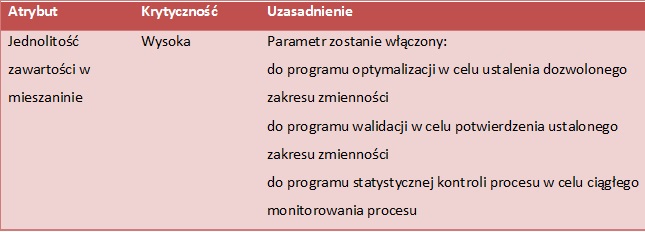
TAB. 8 Ocena strategii kontroli procesu
Zarządzanie ryzykiem jakości
Zgodnie z wymaganiami zaktualizowanego Aneksu 15 zarządzanie ryzykiem jakości zostało wprowadzone do programu walidacji jako usystematyzowany proces oceny, komunikacji i przeglądu kontroli ryzyka w jakości produktu leczniczego w czasie jego cyklu życia. Ocena krytyczności i analiza ryzyka powinna być prowadzona na każdym etapie cyklu życia walidacji. Metodologia oparta na różnicowaniu poziomów krytyczności i ryzyka umożliwia:
- Zmapowanie procesu i identyfikację obszarów szczególnie wrażliwych / podatnych na zmienność a tym samym obciążonych wysokim ryzykiem.
- Identyfikację i ocenę krytyczności atrybutów jakościowych produktu na skali szkodliwości, będącej miarą wpływu atrybutu na skuteczność i bezpieczeństwo stosowania.
- Identyfikację i ocenę krytyczności parametrów procesu na skali, będącej kombinacją stopnia wpływu na atrybuty jakościowe i krytyczności atrybutu jakościowego.
- Ocenę ryzyka procesu na skali, będącej kombinacją szkodliwości atrybutu jakościowego, prawdopodobieństwa wpływu na atrybut jakościowy i wykrywalności.
- Ustalenie priorytetów w programie optymalizacji parametrów, zależnie od poziomu ryzyka i krytyczności w celu scharakteryzowania przestrzeni projektowej.
- Zdefiniowanie odpowiedniej strategii kontroli procesu w zależności od stopnia krytyczności parametrów tak aby zapewnić kontrolę nad zmiennością w procesie.
- Ustalenie atrybutów jakościowych, parametrów materiałów i procesu, które powinny być włączone do programu walidacji.
- Powiązanie zakresu wymaganych aktywności w innych programach systemu zapewnienia jakości (zarządzania zmianami, odchyleniami, reklamacjami) z krytycznością i ryzykiem.
- Identyfikację parametrów materiałów i procesu oraz atrybutów jakościowych, które powinny być włączone do statystycznej kontroli procesu podczas bieżącej weryfikacji procesu.
Ze względu na złożoność procesów technologicznych, które mogą obejmować kilkadziesiąt wzajemnie ze sobą powiązanych parametrów, mogących wpływać na krytyczne dla jakości atrybuty produktu, pełne poznanie produktu i procesu na wczesnych etapach cyklu życia walidacji może nie być możliwe. Konieczne jest więc stosowanie zarządzania ryzykiem jakości jako narzędzia do ustalenia priorytetów opartych na szacowaniu poziomu krytyczności i ryzyka, w celu systematycznego budowania wiedzy o procesie i produkcie w trakcie całego cyklu życia produktu oraz ciągłego doskonalenia procesu poprzez identyfikację, redukcję i kontrolę źródeł zmienności.
Literatura
- EMA, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation (1 October 2015).
- FDA, Guidance for Industry, Process Validation: General Principles and Practices, Revision 1 (Rockville, MD, January 2011).
- ICH, Q8(R2) Harmonized Tripartite Guideline, Pharmaceutical Development, Step 4 version (August 2009).
- ICH, Q9 Harmonized Tripartite Guideline, Quality Risk Management (June 2006).
- ICH, Q10 Harmonized Tripartite Guideline, Pharmaceutical Quality System (April 2009).
- ICH, Quality Implementation Working Group Points to Consider (R2), ICH-Endorsed Guide for ICH Q8/Q9/Q10 Implementation (6 December 2011).
- ISPE, Product Quality Lifecycle Initiative (PQLI) Good Practice Guide, Overview of Product Design, Development, and Realization: A Science- and Risk-Based Approach to Implementation (October 2010).
- ISPE, Product Quality Lifecycle Initiative (PQLI) Good Practice Guide, Part 1-Product Realization using QbD, Concepts and Principles (2011).
- Parenteral Drug Association, Technical Report 60, Process Validation: A Lifecycle Approach (2013).
- Mitchell, M., ‘Determining Criticality-Process Parameters and Quality Attributes’, BioPharm International, Volume 26, Issue 12, December 1, 2013.
- A Recommended Model for Risk-Based Inspection Planning in the GMP Environment, PIC/S Document No. PI-037-1, January 2012.
- EMA-FDA Pilot Program for Parallel Assessment of Quality-by-Design Applications: Lessons Learnt and Q&A Resulting from the First Parallel Assessment, EMA Document No. EMA/430501/2013, 20th August 2013.
Fot. 123rf
